- Install requirements
sudo apt-get install pdfjam texlive-extra-utils samtools bedtools bcftools
- Get reference sequence
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/GRCh38_full_analysis_set_plus_decoy_hla.fa
- Get human sequence data from https://www.internationalgenome.org/data-portal/sample for example HG01504
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/data/IBS/HG01504/alignment/HG01504.alt_bwamem_GRCh38DH.20150718.IBS.low_coverage.cram
- Convert to BAM
samtools view -T GRCh38_full_analysis_set_plus_decoy_hla.fa -b -o HG01504.alt_bwamem_GRCh38DH.20150718.IBS.low_coverage.bam HG01504.alt_bwamem_GRCh38DH.20150718.IBS.low_coverage.cram
samtools index HG01504.alt_bwamem_GRCh38DH.20150718.IBS.low_coverage.bam
-
Find coordinates for TP53 from Ensembl https://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000141510;r=17:7661779-7687538
-
Extract region
samtools view -b HG01504.alt_bwamem_GRCh38DH.20150718.IBS.low_coverage.bam chr17:7661779-7687538 > TP53.bam
samtools faidx ./data/reference/GRCh38_full_analysis_set_plus_decoy_hla.fa chr17:7661779-7687550 > TP53.fasta
We can use bedtools to convert the BAM file to FASTQ. This command will generate two FASTQ files (TP53_R1.fastq and TP53_R2.fastq) for paired-end data. If your data is single-end, only the -fq file will be populated.
samtools fastq -1 TP53_R1.fastq -2 TP53_R2.fastq TP53.bam
- Download VCF
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/ALL.chr17.shapeit2_integrated_snvindels_v2a_27022019.GRCh38.phased.vcf.gz
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/ALL.chr17.shapeit2_integrated_snvindels_v2a_27022019.GRCh38.phased.vcf.gz.tbi
- Filter TP53
bcftools view -r 17:7661779-7687550 -s HG01504 ALL.chr17.shapeit2_integrated_snvindels_v2a_27022019.GRCh38.phased.vcf.gz > TP53.vcf
- Update references (17 -> chr17)
# rename.txt
17 chr17
bcftools annotate --rename-chrs ./data/genes/TP53/rename.txt -Ov -o ./data/HG01504/genes/TP53/TP53_ref.vcf ./data/HG01504/genes/TP53/TP53.vcf
chr17 7661779 7687550 TP53

python gen_mut_model.py \
-r ./data/reference/GRCh38_full_analysis_set_plus_decoy_hla.fa \
-m ./data/HG01504/genes/TP53/TP53_ref.vcf \
-o ./models/mut_model
python genSeqErrorModel.py \
-i ./data/HG01504/genes/TP53/TP53_R1.fastq \
-i2 ./data/HG01504/genes/TP53/TP53_R2.fastq \
-o ./models/seq_error_model
python gen_reads.py \
-r ../data/genes/TP53/TP53.fasta \
-m ../models/mut_model.pickle.gz \
-e ../models/seq_error_model.pickle.gz \
-R 126 \
-o ../data/synth/reads/TP53 \
--bam \
--vcf \
--pe 300 30
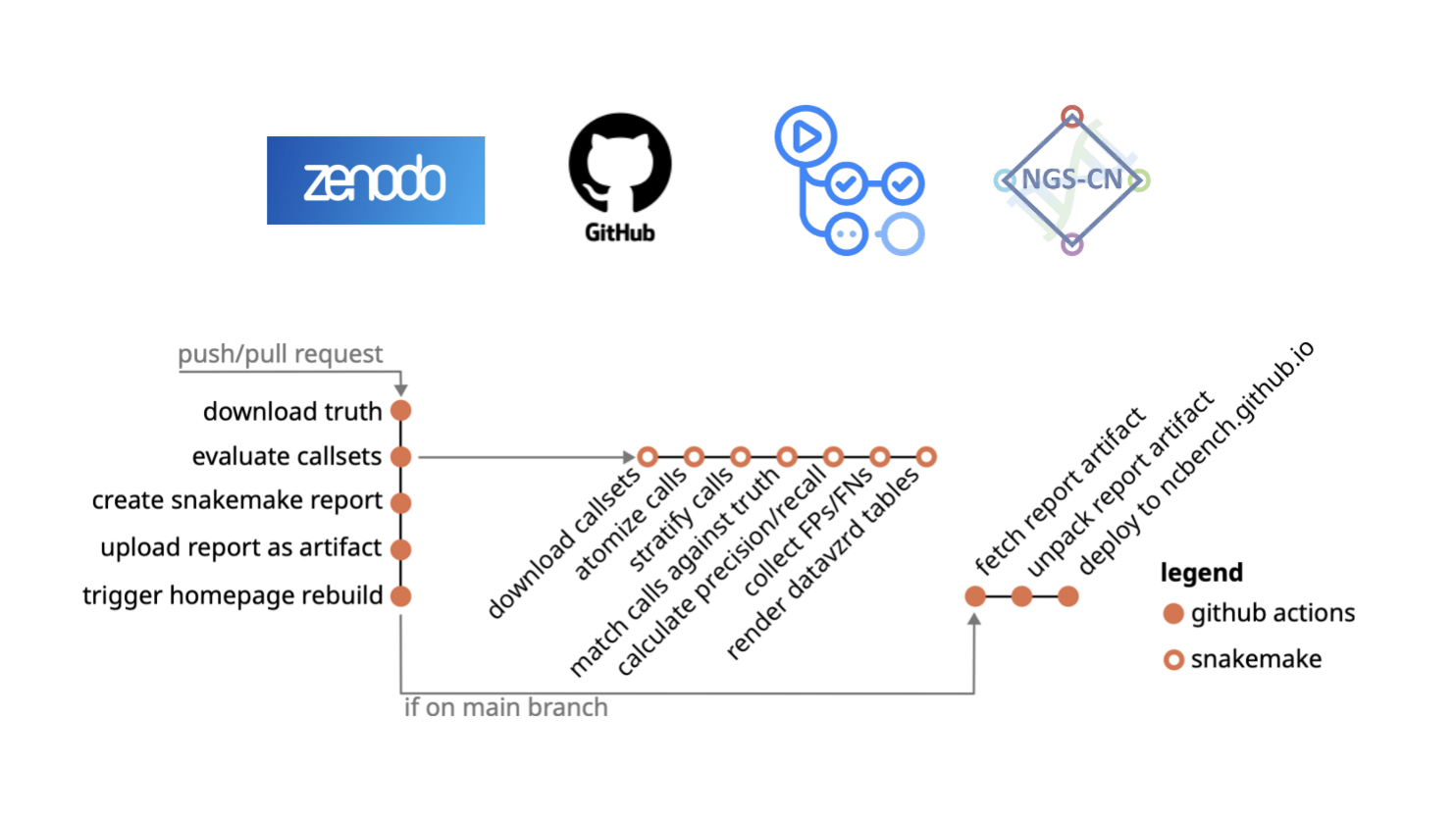
- Check proposed workflow for benchmarking https://www.ghga.de/news/detail/benchmarking-genomic-variant-calling I guess the code to run it is this one: https://github.com/snakemake-workflows/dna-seq-benchmark/