Metagenome comparison toolkit. The toolkit is being developed for EDGE platform and reflects its backend specificity.


install.packages("devtools")
library(devtools)
install_github('seninp-bioinfo/MetaComp')
to use the library, simply load it into R environment:
library(MetaComp)
the_gottcha_assignment <- load_gottcha_assignment(data_file_g)
the_kraken_assignment <- load_kraken_assignment(data_file_k)
the_metaphlan_assignment <- load_metaphlan_assignment(data_file_m)
The package functions load_xxx_assignments (where xxx stands for gottcha, kraken, or metaphlan) are designed to read a tool-specific assignment files. The configuration file for these functions must be tab-delimeted two columns file where the first column is the project id (used as the project's name in plotting), and the second column is an actual assignment file path:
the_assignments_list_g <- load_gottcha_assignments(config_file_g)
the_assignments_list_k <- load_kraken_assignments(config_file_k)
the_assignments_list_m <- load_metaphlan_assignments(config_file_m)
The merge_xxx_assignments function is capable to merge a named list of GOTTCHA, Kraken, or MetaPhlAn assignments into a single table using LEVEL and TAXA columns as ids.
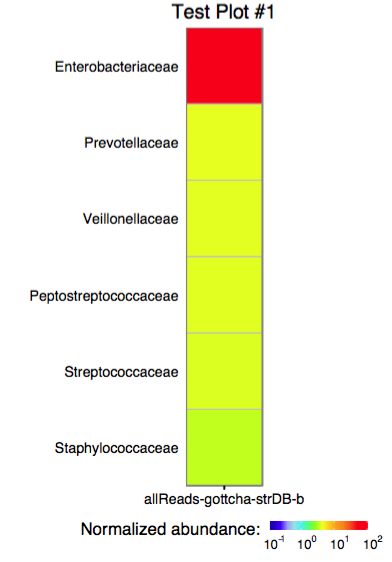
The function plot_xxx_assignment accepts a single assignment table and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
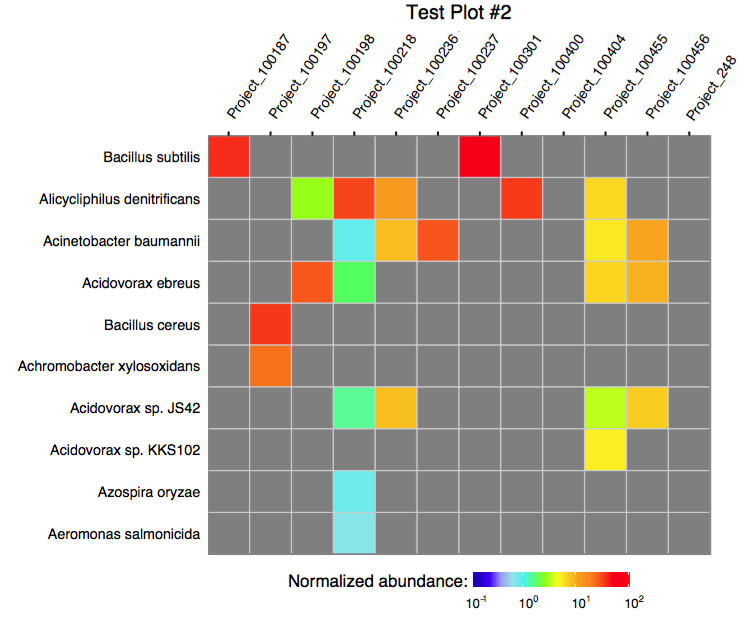
The function plot_merged_assignment accepts a single merged assignment table as an input and outputs a ggplot object or produces a PDF plot using ggplot2's geom_tile.
The following script can be used to run the merge procedure in a batch mode:
# load library
require(MetaComp)
#
# configure runtime
options(echo = TRUE)
args <- commandArgs(trailingOnly = TRUE)
#
# print provided args
print(paste("provided args: ", args))
#
# acquire values
srcFile <- args[1]
destFile <- args[2]
taxonomyLevelArg <- args[3]
plotTitleArg <- args[4]
plotFileArg <- args[5]
rowLimitArg <- args[6]
sortingOrderArg <- args[7]
#
# read the data and produce the merged table
merged <- merge_gottcha_assignments(load_gottcha_assignments(srcFile))
#
# write the merge table as a TAB-delimeted file
write.table(merged, file = destFile, col.names = T, row.names = F, quote = T, sep = "\t")
#
# produce a PDF of the merged assignment
plot_merged_assignment(assignment = merged, taxonomy_level = taxonomyLevelArg,
sorting_order = sortingOrderArg, row_limit = base::strtoi(rowLimitArg),
plot_title = plotTitleArg, filename = plotFileArg)
To execute the scrip, use Rscript as shown below:
$> Rscript merge_and_plot_gottcha_assignments.R assignments_table_gottcha.txt merged_assignments.txt \
family "Merge test plot" merge_test 20 alphabetical
this command line arguments are (some of these are clickable -- so you can see examples):
Rscript- a way to execute the R scriptmerge_and_plot_gottcha_assignments.R- the above script filenameassignments_table_gottcha.txt- the tab delimeted table of assignments (two columns:project_idTABassignment_path)merged_assignments_gottcha.txt- the tab-delimeted output file namefamily- a LEVEL at which the plot should be produced"Merge test plot"- the output plot's titlemerge_test- the output plot filename mask,".pdf"and".svg"files will be produced...20the max number of rows to plot (in the specified sorting order)alphabeticalthe merged plot sorting order
{kind=link}